Acknowledgements
This tool was developed with generous support from the W. M. Keck Foundation and Northwestern University. Partial support from the National Institute of General Medical Sciences (NIGMS) assisted in the creation and maintenance of this website.
Publication
A description of this tool along with several descriptive examples was published in Nature Methods (http://www.ncbi.nlm.nih.gov/pubmed/26780093).
ASMS 2016 Presentation
This video will walk you through the development and application of the Search Engine for Multi-Proteoform Complexes. This tool was designed to be used for the characterization of protein complexes analyzed by native mass spectrometry. Our platform supports a hierarchical search based on a three-tiered tandem MS process. The data that is required for a search includes the intact protein complex mass (MS 1), ejected monomer mass (MS 2), and masses from fragmentation of each monomer (MS 3). This search tool can be used for targeted analysis (purified) or for discovery (endogenous), including the characterization of novel protein complexes. The Search Engine for Multi-Proteoform Complexes is a web-based search tool that is available at http://complexsearch.kelleher.northwestern.edu/.
This presentation was given by Dr. Nicole A. Haverland at the 64th conference on Mass Spectrometry and Allied Topics in June 2016.
Abstract
Efforts to map the human protein interactome have resulted in information about hundreds to thousands of multi-protein assemblies housed in public repositories, but the molecular characterization and stoichiometry of their protein subunits remains widely unknown. Here, we combine the CORUM and UniProt databases to create candidates for an error-tolerant search engine designed for hierarchical top-down analyses, identification, and scoring of multi-proteoform complexes by native mass spectrometry.
Example
Below is an example of the type of results that can be used with this tool. We use the homotetrameric pyruvate kinase complex from rabbit. A summary of the results for this multi-proteoform complex can be downloaded here:

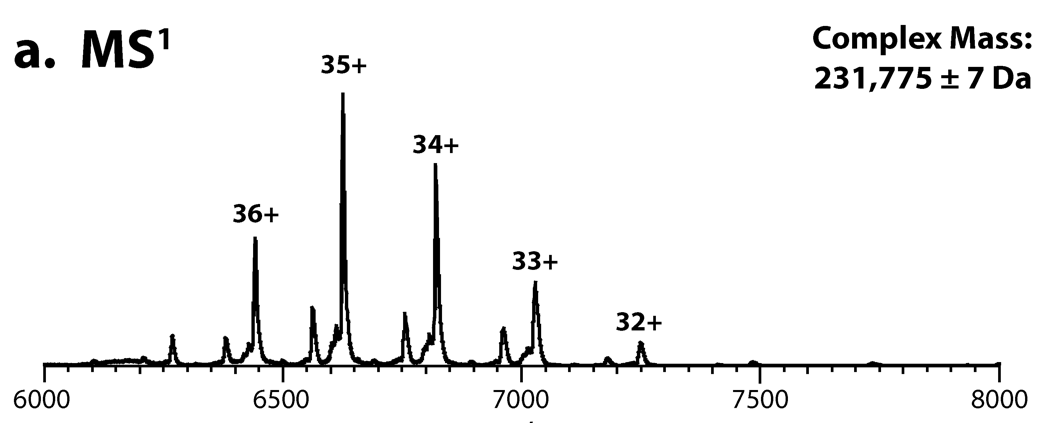
A native mass spectrum of the intact protein complex. The neutral, average MS1 mass can be determined from the most abundant charge states.
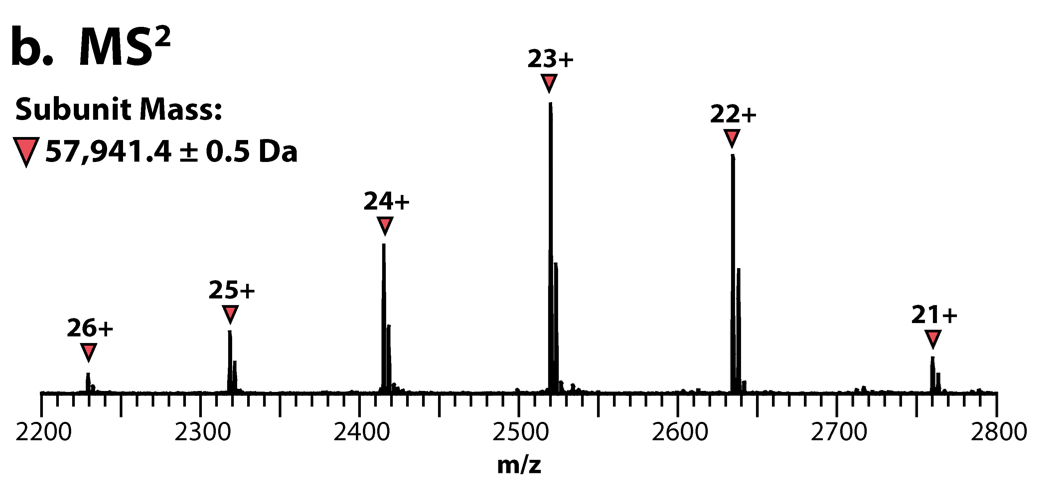
A spectrum of the ejected monomer (subunit). The neutral, average MS2 mass can be determined from the most abundant charge states.

A graphical fragment ion map indicating which fragments have been matched. This map was created by using the deconvoluted, monoisotopic neutral masses from collisionally activated dissociation of the isolated subunit.